What is Galactosialidosis?
Deficiency of an enzyme
Galactosialidosis is a rare genetic disorder characterized by the deficiency of an enzyme called protective protein/cathepsin A (PPCA). This enzyme is involved in the normal functioning of lysosomes, which are cellular structures responsible for breaking down various substances.
Galactosialidosis is inherited in an autosomal recessive manner, meaning that an affected individual inherits two copies of the mutated gene, one from each parent.
The deficiency of PPCA leads to the accumulation of certain substances, including glycoproteins and glycolipids, within the lysosomes. This accumulation affects multiple organs and tissues in the body, resulting in a wide range of symptoms. The severity of the condition can vary widely, even among affected individuals within the same family.
Symptoms
The symptoms of galactosialidosis can include developmental delays, intellectual disability, skeletal abnormalities, coarse facial features, enlarged liver and spleen, heart valve abnormalities, muscle weakness, and seizures. Infants with the disorder may have a failure to thrive and may experience an enlarged head size. Other features that can be present include eye abnormalities, hearing loss, and respiratory difficulties.
Diagnosis
Galactosialidosis is diagnosed through a combination of clinical evaluation, genetic testing, and enzyme activity assays. There is currently no cure for galactosialidosis, and treatment focuses on managing the symptoms and improving the individual’s quality of life. This may involve a multidisciplinary approach, including supportive therapies such as physical therapy, occupational therapy, and speech therapy. Genetic counseling is also an important aspect for affected individuals and their families.
Future
The long-term outlook for individuals with galactosialidosis can vary depending on the severity of the condition and the specific symptoms present. Some affected individuals may have a more severe form of the disorder that leads to a shortened lifespan, while others may have a milder form with a better prognosis. Research and advancements in understanding the condition are ongoing, which may lead to improved treatments or interventions in the future.
We are currently working on several treatment options. We are very optimistic that a treatment will be available soon, so watch this space.
Related Diseases
Sialidosis: Sialidosis is a rare genetic disorder caused by a deficiency of the NEU1 enzyme, leading to the buildup of toxic substances in cells. It has two types: Type 1 (milder, late-onset), which affects movement and vision, and Type 2 (severe, early-onset), which includes developmental delays and organ involvement. There is currently no cure, but research is ongoing.
The therapy for Sialidosis will also treat Galactosialidosis patients so we are closely monitoring the progress in this area.
Please visit curesialidosis.org to find out more about Sialidosis.
Milestones in Galactosialidosis
1968
Initial Clinical Biochemical Identification
1974
Juvenile Patient Identified
1981
Cross Correction in Fibroblast
1982
Molecular identification by D’Azzo
1988
Cathepsin A Cloned
1991
Enzymic Activity of Cathepsin A itself Shown
2012
D’Azzo et al using AAV 2/8 PCCA Murine Correction
2021
PCCA ERT in Murine Model
2024
Submitted pre-IND for AAV gene therapy
2024
Promising Response received from FDA in January 2024
March 2024
Funding applications submitted to start the production of AAV
Classification by Phenotype
Affecting individuals from infancy to adulthood
Early Infantile (10-15%)

Rapidly progressive disease
Cardiac failure
Renal failure
Death before the age of 1
Clinical manifestations
-
- Fetal Hydrops
- Progressive Renal Disease
- Hepatosplenomegaly
- Splenic Sequestration
- Hernias
- Telangiectasia
Late infantile (5-10%)

Dysostosis multiplex
Spine / Thorax
Renal failure (rare)
Hepatosplenomegaly
Cardiac involvement
Hearing Impairments
Visual Impairements
Dentition
Growth Issue
Height Issue
Arthritis (one case)
Limited CNS manifestations
Juvenile/adult (80%)
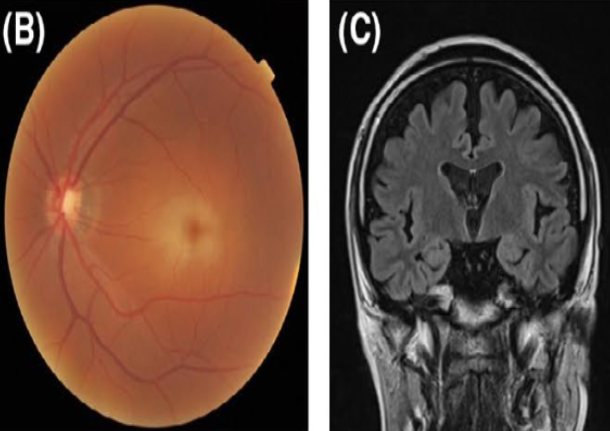
Predominate Neurological
Myoclonus
Ataxia
Seizures
Mental retardation
Visual impairment
Angiokeratomas
Approx. 150 Patients in the World
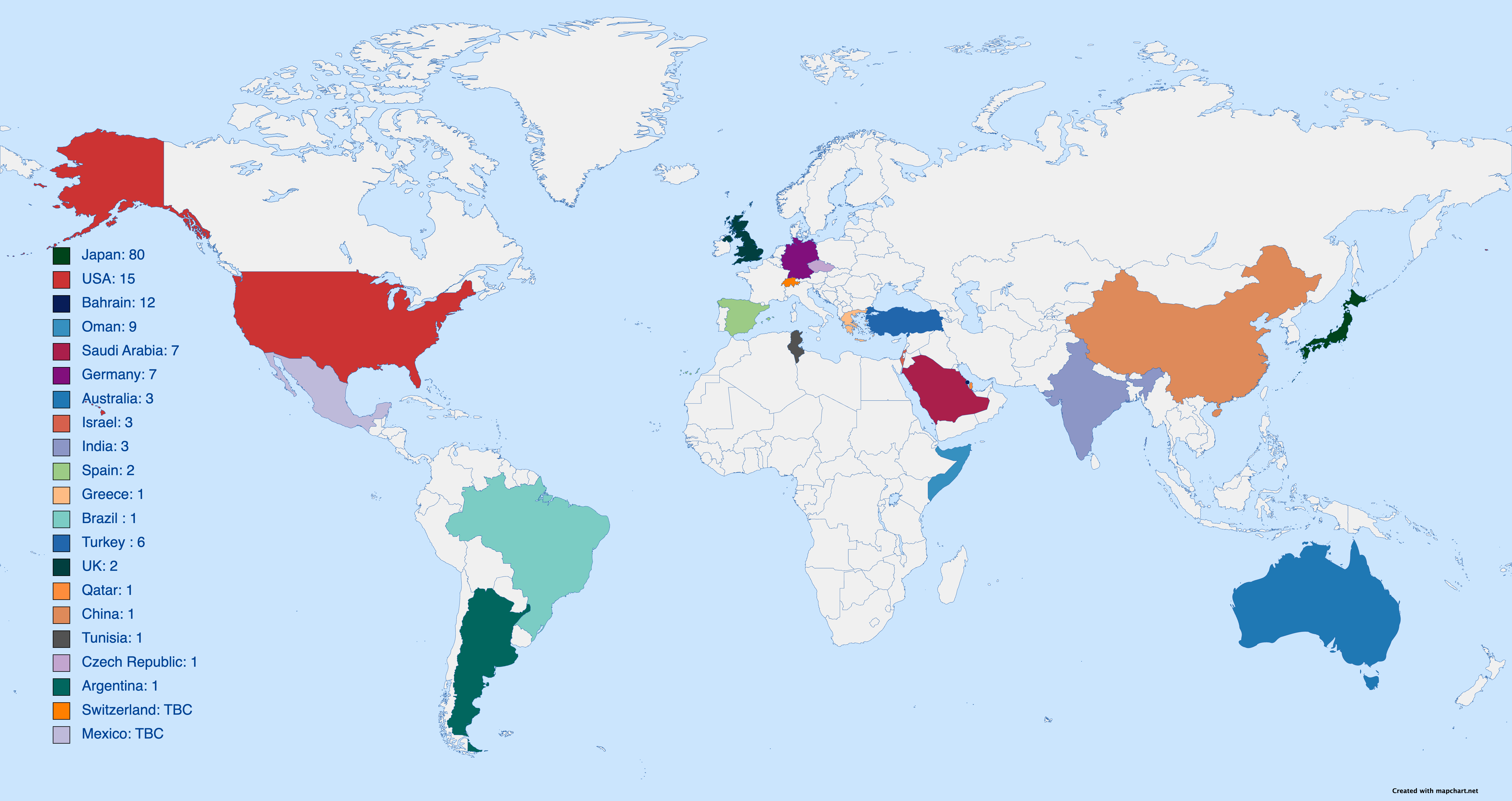
| Country | Living | Deceased | Total |
| Switzerland (TBC) | – | – | – |
| Mexico (TBC) | – | – | – |
| Japan | – | – | 60 |
| USA | 12 | 3 | 15 |
| Bahrain | 7 | 5 | 12 |
| Oman | – | – | 9 |
| Saudi Arabia | – | – | 7 |
| Germany | – | – | 3 |
| Turkey | 1 | 5 | 6 |
| Australia | – | – | 3 |
| Israel | – | – | 3 |
| India | – | – | 3 |
| Spain | 2 | 0 | 2 |
| UK | 1 | 1 | 2 |
| Greece | 0 | 1 | 1 |
| Brazil | – | – | 20 |
| Qatar | 1 | 0 | 1 |
| China | 0 | 1 | 1 |
| Tunisia | – | – | 1 |
| Czech Republic | 0 | 1 | 1 |
| Argentina | 1 | 0 | 1 |

Get in touch
If you would like more info or would like to collaborate with us to help find a treatment, please contact us using the details below:
You can also join the ISMRD mailing list.
International Directory of Clinicians and Scientists with experience of galactosialidosis are cited here, if you are a patient, parent or professional with an interest in galactosialidosis you may want to get in touch.
